Conformer generation
Edited: update the figure after working around the RMSD alignment (rdkit tries all the atom order permutations to find the best alignment, rather than keep the atom order fixed, i think); removed the technical problems paragraph. New figure.
“Conformer generation is an art”. That statement from a more senior computational chemist initially made me chuckle – I mean, how hard could that be? If computational methods are claiming to bring a lot to the table, surely conformer generation is a problem solved long ago… Oh the naive young mind ;)
Let’s start with a simple question here is: if we start from the native (experimentally observed) conformation of a ligand and minimize it using two popular ligand force-fields (UFF and MMFF), we should stay close to the crystal structure. Is that indeed the case?
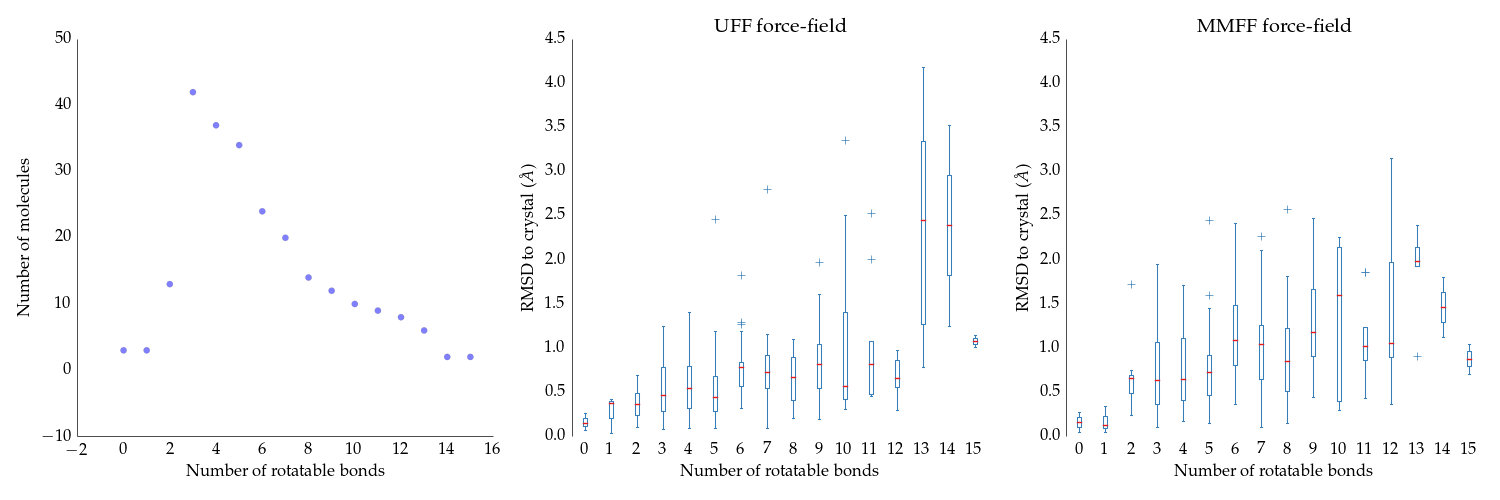
Figure 1
So that doesn’t look so bad! Let’s hydrogenate the structures – right now they’re mixed bag.

Figure 2
Woops, adding hydrogens shouldn’t increase the number of rotatble bonds (at least not in the conventional sense).
MMFF appears to be doing better for ligands with large number of rotatable bonds – however, we have relatively low number of those. For ligands with 4-6 rotatable, which constitute most of the dataset, the UFF seems to be doing better. So the seeming advantage of MMFF may come just from the fact that we have a small number of compounds with large number of rotatable bonds.
Neither MMFF nor UFF correctly account for the trans/cis orientation of amides. This may or may not have an impact on this benchmark, depends how many amide-containing compounds are in this set (if there are none, even if correct amide handling is introduced there will be no change to the plots above).
Another question is what if we start from a random conformation (a more real-life problem). How likely would the UFF/MMFF minimization yield a conformer close to what’s observed in a crystal?
The silent assumption here is that the experimentally observed conformers, predominantly from crystal structures, are correct. That’s often not true.
If one wishes to, say, write a docking code getting those ‘simple things’ such as conformer generation right is critical. If that’s wrong, even if you have an ‘ideal scoring function’ (if such thing exists…) and an ideal sampling scheme won’t get you all the way.
Many thanks to JP for providing his set, here is JP’s paper on conformer generation.