Measuring the performance of ligand-based methods
This is another episode in the “how good are these methods really?” series. The aim is to understand how well the ligand-based 3D methods for virtual screening perform under standard benchmarking sets. In the original papers introducing the methods frequently is benchmarking done on a small test sets, prone to all sorts of small population problems.
Here I take 3 popular methods: the ROCS-like implementation of Gaussian shape overlap[1] that I recently ported to rdkit as well as USR[2] and USR-CAT[3], two very fast shape methods. All 3 are tested on the DUDE[4] dataset, a standard benchmarking set. Conformer generation was done as outlined by JP in his paper[5].
| Pair of methods compared | Statistic used | Value |
|---|---|---|
| rdkit-shape VS usr | wilcoxon | T=2.24, p=0.02 |
| rdkit-shape VS usrcat | wilcoxon | T=-0.47, p=0.63 |
| usr VS usrcat | wilcoxon | T=-2.70, p=0.006 |
One thing that struck me was the spread of performance. I got a little worried that the conformer generation method was messed up, so I tested that part and it correctly reproduced the crystal poses to within 2-3 Angstrom easily (below). So that part is okay.
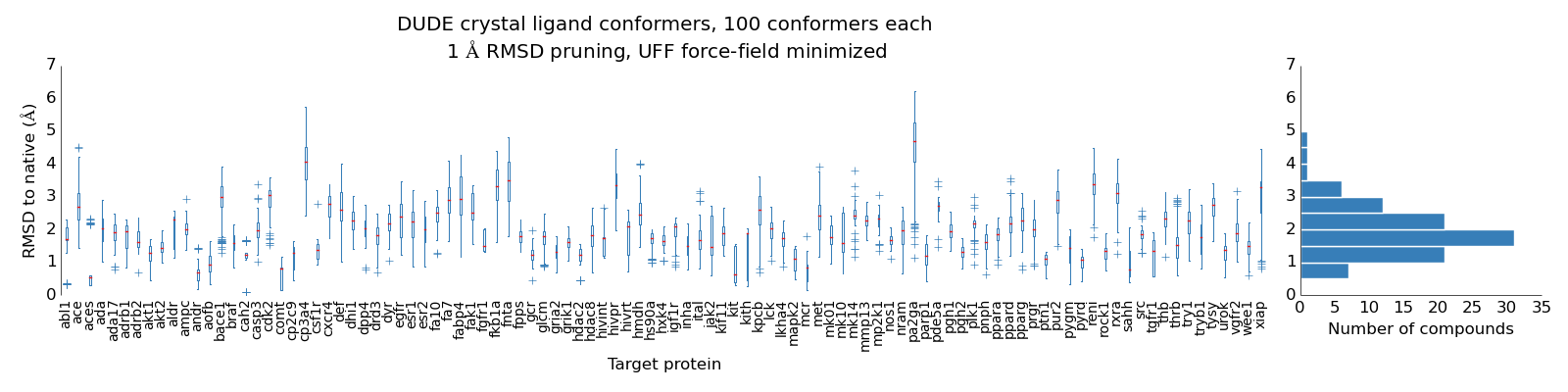
It still could be that there is a numerical problem with rdkit-shapeit – and that it can’t find the correct maximum overlap but I’m not sure how to test for that.
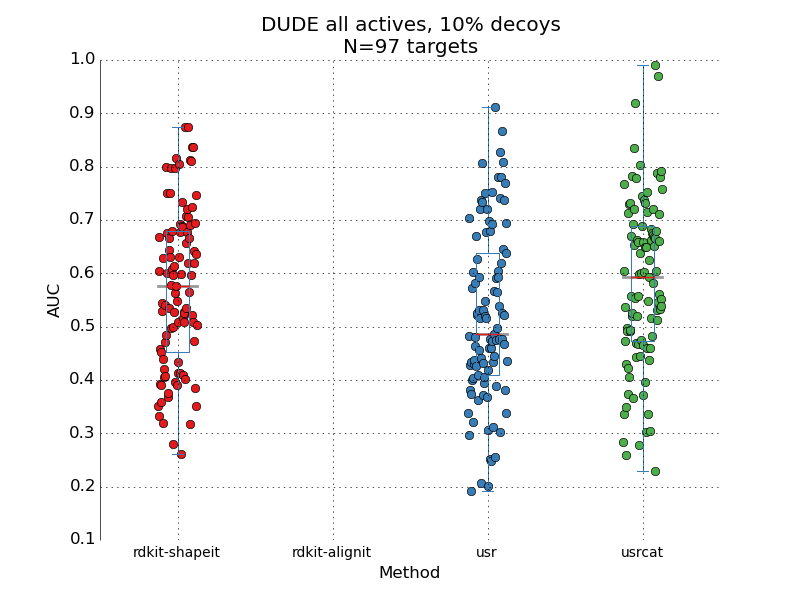
But let’s take it all at a face value. These methods are not great… USR is worst of all, kind of making a case that it doesn’t matter if the method is fast, it matters if it gives any sort of sensible results. rdkit-shapeit isn’t much better. USRCAT performs best of all, along the lines of “if you include some chemistry features, it works much better”. It seems that although shape complimentary is a necessary condition for molecular similarity, it’s certainly not a sufficient condition…
Going back to the general statement (that these methods don’t look awesome), it’s clear that even the best one offers a median AUC (0.6) that’s barely above random (0.5). The interquartile range for all 3 methods is huge, which means that depending on your target protein they may work well or terrible. It’s interesting to think that a method with a narrow interquartile range around AUC of 0.6 would be more desirable that a method with median AUC of say 0.7 but with huge variation from target to target.
A blank space is left for rdkit-alignit, a pharmacophore tool. rdkit-alignit is to rdkit-shape what usrcat is to usr – it incorporates chemistry features into the search. The blank space is left because I haven’t ported alignit into rdkit yet and so I can’t make the comparison.
References: [1] http://onlinelibrary.wiley.com/doi/10.1002/%28SICI%291096-987X%2819961115%2917:14%3C1653::AID-JCC7%3E3.0.CO;2-K/abstract [2] http://www.ncbi.nlm.nih.gov/pubmed/17342716 [3] http://www.jcheminf.com/content/4/1/27 [4] http://pubs.acs.org/doi/abs/10.1021/jm300687e [5] www.ncbi.nlm.nih.gov/pubmed/22482737