gromacs 5.0 - first look
Oh, gromacs, old friend let’s see what you have to offer in your new release
wget ftp://ftp.gromacs.org/pub/gromacs/gromacs-5.0.4.tar.gz
tar xvf gromacs-5.0.4.tar.gz
cd gromacs-5.0.4
mkdir build
cd build
cmake ..
Problem #1:
-- Could NOT find LibXml2 (missing: LIBXML2_LIBRARIES LIBXML2_INCLUDE_DIR)
CMake Warning at CMakeLists.txt:601 (message):
libxml2 not found. Will build GROMACS without unit-tests. This is not
recommended, because the unit-tests help to verify that GROMACS functions
correctly. Most likely you are missing the libxml2-dev(el) package. After
you installed it, set GMX_BUILD_UNITTESTS=ON.
Problem #2:
CMake Warning at cmake/gmxManageFFTLibraries.cmake:94 (message):
The FFTW library was compiled with --enable-avx to enable AVX SIMD
instructions. That might sound like a good idea for your processor, but
for FFTW versions up to 3.3.3, these are slower than the SSE/SSE2 SIMD
instructions for the way GROMACS uses FFTs. Limitations in the way FFTW
allows GROMACS to measure performance make it awkward for either GROMACS or
FFTW to make the decision for you based on runtime performance. You should
compile a different FFTW library with --enable-sse or --enable-sse2. If
you have a more recent FFTW, you may like to compare the performance of
GROMACS with FFTW libraries compiled with and without --enable-avx.
However, the GROMACS developers do not really expect the FFTW AVX
optimization to help, because the performance is limited by memory access,
not computation.
Call Stack (most recent call first):
CMakeLists.txt:738 (include)
Problem #1 is easy, at least on Ubuntu 14.04:
sudo apt-get install libboost-dev libxml2-dev
For Problem #2, we’ll just ask gromacs to compile it’s own FFTW. Additionally the built will be done in single precision with GPU support:
cmake .. -DGMX_GPU=on -DGMX_DOUBLE=off -DGMX_BUILD_OWN_FFTW=ON
make -j40
make install
Warning: i’m not sure if at this stage I need to worry about OpenMP vs MPI. All runs will be single node, to test the GPU code, so I’m going to ignore the MPI, which in my understanding is only used for inter-node simulations?
Good, let’s compile an equivalent version of the gromacs 4.6.7 and compare the two creatures. The expectation is that the performance is not drastically different. The way we measure the performance is by taking a number of water boxes of increasing size.
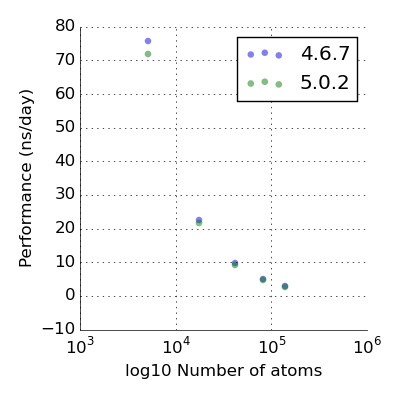
With a single K20 GPU and 40 OpenMP threads, the GPU is grossly overworked. By chemical terminology those are “CPU-saturating conditions”. With that caveat the performance is not mind blowing - for a chemically meaningful system size 10^4-10^5 it’s around 20 ns/day but K20 is not exactly ‘top of the line’.