State of co-folding 2025
Last week, the co-folding world exploded: Pearl, OpenFold3, and BoltzGen all dropped within 48 hours. With so many AF3 “clones” now in play, I wanted to map out who’s who and what challenges these models face in 2026.
Why does structure prediction matter?
Structure prediction is a 2nd order thing to drug-discovery programs, where people really care about:
1. Finding therapeutic targets to go after, 2. finding hit molecules for those targets, and 3. optimizing those hit molecules.
In the discovery context, structure prediction is a foundation, a “means to an end”, not the goal itself – that’s markedly different from a tech crowd, where perhaps “making the number go higher” (benchmarks) is seen as sufficient.
How does structure prediction impact lead optimization? Well, for one thing, better co-folding models equals better starting structures for FEP simulations. As these models improve geometry and chemistry awareness, they’ll reduce the setup time and expert touch currently required for FEP workflows.
Comparisons are tricky
The public structure prediction benchmarks often focus on aspects of the predicted pose, like ligand RMSD to crystal structure. Geometric problems are often incompletely captured but that’s changing: people are looking at non-physical protein/ligand/water contacts and geometries.
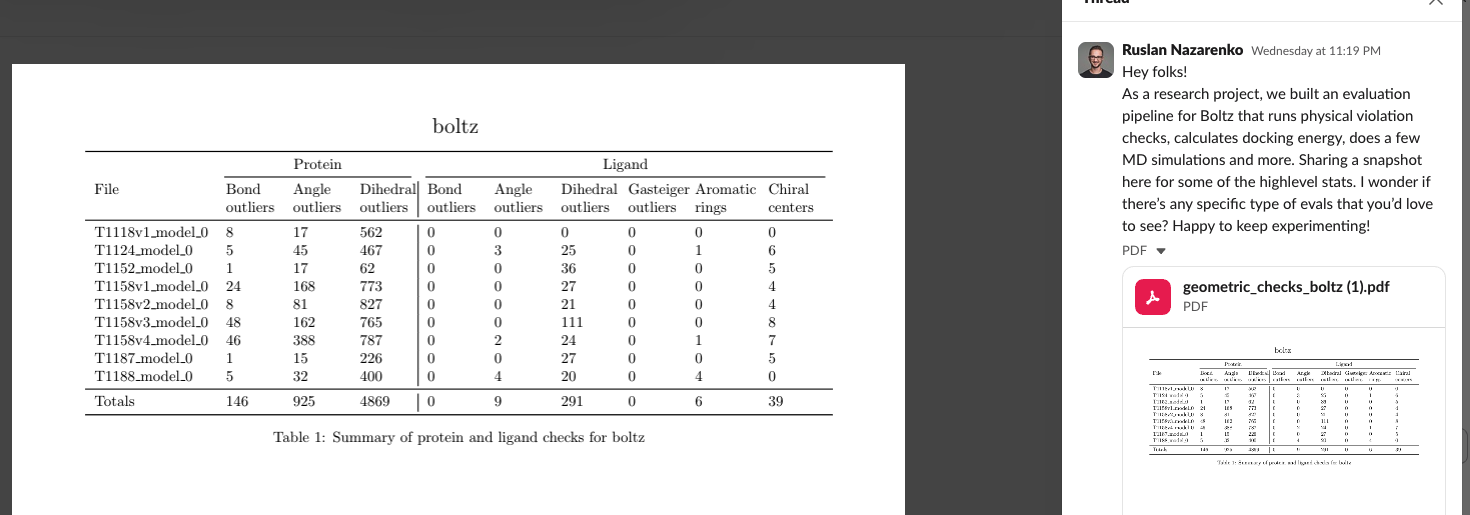

While many SAAS products (Nvidia NIM, Tamarind.bio, Deepmirror, Rowan) will allow you to run these models, almost none of them really allow you to benchmark (Apheris is a notable exception). Simple inference is the level of “kicking the tires”, barely above Hugging Face examples, and hard to imagine how that fits into a discovery workflow.
Who’s who 2025
| Model | Parent company / org | Open source? | What’s special |
|---|---|---|---|
| AlphaFold 3 | Google DeepMind & Isomorphic Labs | ❌ No (free server access for non-commercial use) | “The OG”: universal co-folding, which predicts structures/interactions for proteins, DNA/RNA, small molecules, antibodies, and ions. |
| RoseTTAFold All-Atom | Baker Lab / RoseTTAFold team | ✅ Yes (open GitHub release) | Extends RoseTTAFold to full biomolecular assemblies — proteins plus DNA/RNA, ligands, metals, and covalent modifications |
| Boltz-1 | Boltz (MIT Jameel Clinic collaborators) | ✅ Yes (MIT License; code & weights) | Open all-atom co-folding. |
| Boltz-1x | Boltz | ✅ Yes (MIT) | Boltz-1 variant. |
| Boltz-2 | Boltz | ❔ Weights-only (MIT), no training code, no data | Affinity prediction. |
| BoltzGen | Boltz / MIT & collaborators | ❔ Yes (MIT), no data | Generative peptide/protein model on top of Boltz-2. |
| Chai-1 | Chai Discovery | ✅ Yes (Apache-2.0) | Biologics-only? |
| Protenix (“Protein-X”) | ByteDance | ✅ Yes (Apache-2.0) | - |
| HelixFold3 | Baidu | ❔ Yes, non-commercial only | - |
| NeuralPLexer3 | Iambic Tx | ❌ No (proprietary) | - |
| DragonFold | CHARM Therapeutics | ❌ No (proprietary) | Matches AF3 on small-molecule prediction, used in discovery. |
| Pearl | Genesis Molecular AI | ❌ No (proprietary) | Exceeds AF3 on small-molecule prediction, used in discovery. |
| OpenFold 3 | OpenFold Consortium | ✅ Yes (preview open-source release) | First open-source re-implementation to match AF3 performance across modalities (RNA, small molecule, biologics). |
Favorites for 2026
Closed-source: Pearl
Probably the best closed-source model for small molecules at the moment, released earlier this week. Another giggle: the company renamed from “Genesis Therapeutics” to “Genesis Molecular AI”… I see what you did here! With the (dire) state of the biotech investment, everyone seems to be cuddling closer and closer to “AI”, where investment dollar flows happily.
Excited to share Pearl from Genesis Molecular AI (yes, we've updated our name!): the first co-folding model to clearly surpass AlphaFold 3 on protein-ligand structure prediction.
— Genesis Molecular AI (@genesistxai) October 28, 2025
Unlike LLMs that train on vast public data, drug discovery AI faces fundamental data scarcity. Our… pic.twitter.com/Jmc2FQ65mA
As an ex-competitor of theirs, this is certainly interesting to me: Charm Therapeutics also focused on small molecules. CharmTx DragonFold model was able to match AF3 but not meaningfully exceeded it. The Genesis crew significantly outperforms AF3 on small-molecule structure prediction.
The code and model remains closed-source, even to academics/non-commercial users. The technical report which accompanied the release is sparse on the details but if you’ve worked in this field, the subtle nudges within are pretty clear: some mixture of architecture changes, and synthetic data generation.
The team is suitably cross discipline and appears well funded. Such teams will continue to outperform pure tech plays – in my view – because pure tech will often tap-out in the data curation angle, or success metric definition: the ability to curate data with help of crystallographers, affinity data with biology and chemistry teams, ability to generate synthetic data with computational chemists. “Make number go higher” only works if you have a clue how to define the number, and your science colleagues do.
Open-source: OF3
I personally share the enthusiasm of the post below :D
🔥 It's here: OpenFold3 is now live.
— Georgia Channing (@cgeorgiaw) October 28, 2025
THE open-source foundation model for predicting 3D structures of proteins, nucleic acids & small molecules. This is where the future of drug discovery and biomolecular AI lives.
Built by @open_fold. Hosted on @huggingface.
👇 more pic.twitter.com/IukdhBL8rD
OF3 certainly looks to be more “opener” than “open-weights” competitors. To be fully and meaningfully open, the models should disclose their training code and data. Weights and inference code just allows the model to be run “as is”.
What’s important for OF3, and more broadly the open-model community, is to move beyond AF3 and into the future. The OF3 crew has the (massive) benefit of multiple talents in the house (tech + science, academia + industry) – bringing those together will be the key to success.
Challenges and ideas
Generalization
The problem Performance drops sharply on targets not seen in training. Models interpolate well but struggle to extrapolate.
Why it matters Biologics – where the hypervariable Ab regions by definition don’t have many “similar” sequences in the training dataset. Small molecules – targets with limited structural data.
What could work
- better architectures to increase what the model can squeeze out of the same data,
- federated learning to provide proprietary data,
- better measuring sticks, Runs’N’Poses (RnP) is a great start.
Validation beyond benchmarks
The problem The public structure prediction benchmarks are a “minimum bar” to pass. Most of the AF3 clones have not been tested in discovery.
Why it matters The benchmarks don’t fully capture the expectations of a medicinal chemist, trust is lost when used prospectively and artifacts are observed.
What could work
- Better benchmarks, with more medicinal chemistry focus
- Back-testing on industry data (preferably data not seen in training).
- Blind prospective validation – but that takes time and money.
Protein dynamics (including disorder)
Protein dynamics and protein disorder comes in many different flavors: from fully disordered proteins, to those that fold upon contact with others “partially disordered”. On the more humble end, you could think as allostery as the minimal case of protein disorder albeit that’s a bit unorthodox.
The problem Current generation of models are great “structure predictors” and don’t appear to have a great understanding of protein physics/dynamics, folding mechanism or kinetics/thermodynamics is out of reach.
Why it matters Many drug targets are partially disordered or undergo conformational changes upon binding. Single static structures miss the ensemble.
What could work
- Training on experimental stability/flexibility data (HDX-MS, radical reactivity)
- Synthetic MD trajectories, e.g., AF CALVADOS
Data scarcity and quality
The problem Unlike LLMs trained on the entire internet, structure models are data-starved. Self-distillation risks “exhaust gas recirculation”—training on your own mediocre predictions degrades quality.
Why it matters: More data = better performance, but only if the data is high-quality. Garbage in, garbage out.
What could work
- Synthetic data generation, with careful curation
- Molecular dynamics (Boltz-2)
- From affinity datasets with closely-linked structural data (Genesis?)
- Experimental data generation for structure and affinity (OpenBind consortium)
- Fine-tuning on proprietary data (works better than RAG for these models)
Scaling laws
The problem We haven’t seen clear scaling laws emerge yet, where same architecture can be scaled up to a larger model and unlock new capabilities.
Why it matters Scaling laws drive investment, without them only data/architecture breakthrough are needed.
What we’re learning Genesis hints at scaling with synthetic data. More investigation needed.
Chemistry awareness
The problem Models still generate non-physical geometries – protein and ligand impacted – bad bond angles, steric clashes, impossible ring conformations.
Why it matters Medicinal chemists won’t trust poses that violate basic chemistry. “This nitrogen can’t be sp3 in this fragment” = loss of trust in the tech.
What could work
- Improve the handling of chemistry features: bond orders, protonation, chirality, ring puckering, non-physical intra-molecular contacts,
- Physics-based geometry constraints during training/inference,
- Water co-folding Many discovery programs hinge on explicit water interactions—currently missing from all models.
Affinity prediction
The problem Current affinity models appear to overfit training distributions. Performance drops on novel scaffolds or targets not in training data.
Why it matters In lead optimization, accurate ΔΔG (relative binding affinity) prediction can save money spent on synthesis and experimental validation.
What could work:
- Split the affinity problem into two: (1) binder/non-binder separation between different series, and (2) accurate affinity prediction within a series,
- Synthetic data: pair known affinity datasets with predicted/AlphaFilled structures
Training complexity
The problem Multi-stage training regimes make reproduction difficult. These models aren’t as big as LLMs (64 A100s suffices) but the “fiddly” training process creates replication barriers.
Why it matters Complex training = fewer groups can validate/extend the work, because they have to “guess” the training stage parameters.
Architecture frontiers
The problem Hard to predict what architecture changes will matter. Attention mechanisms have memory limitations for all-atom polymer representations.
Why it matters Current generation of models could be permanently limited because of the loss of all-atom representation, needed to save memory.
What could work
- Pearl hint at the return of SO(3)-equivariant architecture (present in AF2 but removed in AF3)
- Improved coarse-graining: 2-3 tokens per residue instead of 1 (captures more geometry without full all-atom overhead)
- Hybrid approaches: coarse for bulk structure, all-atom for binding site (already the case for PTM residues, which are all-atom)
Why do inverse folding methods hate bulky aromatics so much? From the BoltzGen paper pic.twitter.com/BhDcOHuk42
— Diego del Alamo (@DdelAlamo) October 27, 2025
Conclusion and predictions for 2026
This field is currently pretty buzzing or at the very least showing “healthy signs of activity”. These models are here to stay, and are quickly becoming the bread-and-butter of discovery. We understand much better where they work and why. However, there are quite of few of them and the differences are to express (commodification at least at the surface level).
Maybe we’ll cure all diseases in 10 years – that’d be really awesome – but we probably won’t. We need more AF3-like moments for different parts of the drug discovery process.
Prediction is hard, I was certainly wrong about two major things last year
- I was skeptical of federated learning, yet that strategy seems to be doing pretty well (eg Apheris, OpenFold)
- I was skeptical of synthetic data via but that too seems to have done well (Genesis and Boltz have reported on this, albeit have taken different strategies)
For 2026, I think we’ll see multiple models go beyond AF3, both in terms of capabilities and raw structure prediction performance. People are definitely trying to address the data limitations, both via synthetic data generation and experimentally via initiatives like OpenBind. Targets with little structural data will remain challenging, unless new break-through architectures emerge.
Scientists, talk to your tech buddies – tech people talk to your science mates. None of you can do it alone, only together can you complete the puzzle.
Acknowledgements
Many thanks to Peter Mernyei & Jenke Scheen for their feedback on this post.